Bone loss in AS
It is now well established that patients with AS are at high risk of developing osteoporosis (Davey-Ranasinghe 2013). The prevalence of osteoporosis and osteopenia in ankylosing spondylitis, as assessed by cross sectional studies, varies from 2- 34% and 26-52 % respectively (Table 3). Thus, it is evident that osteoporosis is an extra articular manifestation that has a prevalence similar to or even higher than that of other well-known extra articular manifestations of AS such as iritis, IBD and psoriasis (El Maghraoui 2011). Despite being common, bone loss is seldom suspected and investigated, because of young age and predominantly male gender of the affected cohort.
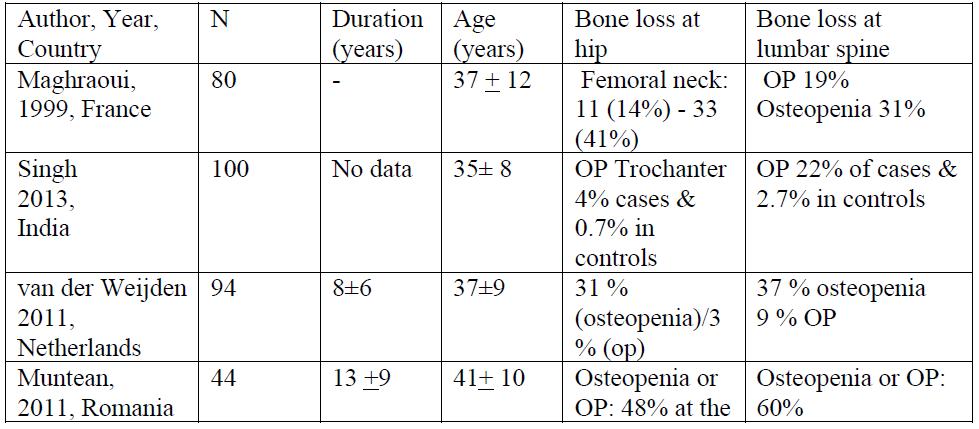
Table 3: Prevalence of bone loss in patients with AS

Table 3: Prevalence of bone loss in patients with AS
Data explaining the prevalence of bone loss in AS should be interpreted with caution as bone loss in AS is influenced by many factors. First, many studies have overestimated the prevalence rates as a consequence of applying WHO definition to define osteoporosis and osteopenia. However, it is now clear that the WHO definition cannot be used to define bone loss in young males and pre-menopausal women. This is because using the WHO criteria in young subjects overestimates the prevalence of bone loss. Thus, the ISCD recommends using Z scores to define bone loss in men aged less than 50 and premenopausal women. A cross sectional study done by Vasdev and colleagues addressed this issue (Vasdev 2011). The authors tried to analyze the prevalence of bone loss in AS patients who were younger and hadshorter duration of disease than the prior studies. The patients were aged 33+ 8 years and had had AS for 8 ± 6years. In this case control study conducted in India, osteoporosis at the spine and femur neck was present in 29% (controls: 2%, p < 0.001) and 11% (controls: 1%, p < 0.001), respectively. The observed prevalence was lower when bone loss was defined using Z scores: low bone density in spine and femur was seen in 24% (controls: 1%, p < 0.0001) and 5% (controls: 0%, p < 0.01), respectively. Second, the prevalence rates of bone loss depend based on the site of measurement of BMD, as the extent of bone loss at the lumbar spine and hip are different. Osteoporosis and osteopenia at the spine have been reported in 13-34 and 20- 44% of patients respectively. Similarly at the hip, 2-33 and 26-52% of AS patients have been found to have osteoporosis and osteopenia at total hip or femoral neck. Third, the prevalence rates of bone loss also differ based on the technique used for DXA imaging.
For instance, lateral spine BMD is likely to be more accurate than anteroposterior spine BMD in AS (Borman 2008, Klingberg 2012, Ulu 2013, Bronson 1998). In a study done by Borman and colleagues, DXA of lateral spine was used to study bone loss (Borman 2008). In this cross sectional case-control study, there were 32 AS patients with mean disease duration of 15 years and osteoporosis was observed in 11 (34%) AS patients (vs. 6% in controls). The prevalence of bone loss in AS is also influenced by gender. Osteoporosis in AS affects both men and women but bone loss is less common in women with AS compared to men. Men are thought to have more severe disease than women. The gender differences in bone loss may be due to the protective hormonal effects in premenopausal women or may be due to women having less severe disease (van der Weijden 2011). In women, menopause is an important accelerating factor for bone loss. Thus, Muntean and colleagues decided to analyze the prevalence of bone loss in men alone so that confounding effects of gender was eliminated (Muntean 2011). Men with AS (n=44) who were aged less than 60 were eligible to participate in this cross sectional study. The subjects had a mean age of 41+ 10 years and disease duration of 13 +9 years. BMD at femoral neck and total hip (.90 vs. 1.03 in controls and .94 vs. 1.05 in controls) were lower in men with AS compared to controls (both p =0.001). Lumbar spine BMD was also lower in patients compared to controls, but the difference did not reach significance (1.16 vs. 1.06, p = 0.06). This study observed a greater prevalence of bone loss than was previously reported.
Osteopenia or osteoporosis was found in 60% of AS patients at the lumbar spine and in 48% at the femoral neck. Finally, the nature of bony involvement of the spine in AS may also influence the prevalence of bone loss. Lumbar spine DXA is likely to document normal values in patients with bony changes at spine and consequently underestimate bone loss (Arends 2011). Karberg and colleagues demonstrated this by categorizing patients based on their disease duration. Osteoporosis at the hip and spine were found by DEXA in 11% and 15%, respectively, in 27 patients with early AS (duration < 5 years). Lumbar spine DXA was abnormal in only 4% of patients who had prolonged disease (> 10 years) as opposed to 11% in those who had shorter disease duration. More patients were found to have osteoporosis at the hip (29%) with long standing disease.
Data on longitudinal bone loss in AS
Not much is known about whether bone loss in AS occurs in a progressive manner. Most of the existing evidence on bone loss in AS is based on data obtained from cross sectional studies. This limits our ability to make conclusions regarding a causal association. Thus far, only three studies have assessed longitudinal changes of BMD in AS patients who were treated with conservative treatment (Kang 2011,Maillefert 2001, Kaya 2009). The maximum duration of follow up in these studies was two years. Longitudinal data suggest that after two years of follow up, BMD at lumbar spine either remained stable or increased. Conversely, BMD at the hip either remained stable or declined over a period of two years. In a retrospective longitudinal study conducted in Korea, the lumbar spine BMD increased from 1.17+ 0.15 to 1.20+0.16, p=0.01 (baseline vs. year 1), but femoral neck BMD did not change significantly (0.98+ 0.15 vs. 1.00+ 0.15, p=0.34 (Kang 2011). The subset of patients analyzed had been treated with conventional measures and NSAIDs. However, the sample size was very small. It is also likely that patients, who were treated with NSAIDs alone, had milder disease and bone loss at hip might take longer than one year to manifest. In another longitudinal study conducted in Turkey (n=55, mean age =36 +11 years, duration= 11+ 8 years) the lumbar spine BMD was noted to increase by 3.4% over 24 months. In contrast, BMD at femoral neck and total hip decreased by 0.9% and 0.25% after 24 months. In the sub group analysis, percentage changes in BMD measurements were not significantly different between patients with active (n = 10) or inactive (n = 11) disease. In another study, Maillefert and colleagues examined the longitudinal changes in BMD over two years (Maillefert 2001). Fifty-four patients aged 37 + 11 years (disease duration: 12.4 + 8.6 years) were followed up for two years. After 2 years, BMD at the lumbar spine did not change (+0.75% +3.5, p = 0.23). But significant bone loss was demonstrated at the femoral neck (–1.6%+4, p = 0.006). QCT was used by Korkosz and
colleagues to demonstrate longitudinal bone loss in AS (Korkosz 2011). In this small study of 15 patients, the authors found that trabecular bone mineral content at the spine measured by QCT, decreased significantly after ten years (change ± SD: 18 ± 7 mg/![]() , p=0.001). DXA at the spine revealed a significant increase of BMD (change ± SD: -0.15 ± 0.14 g/
, p=0.001). DXA at the spine revealed a significant increase of BMD (change ± SD: -0.15 ± 0.14 g/![]() ) in contrast to the results of QCT. So far, no other study has attempted to replicate these results.
) in contrast to the results of QCT. So far, no other study has attempted to replicate these results.
Risk factors of bone loss in patients with AS
Bone loss in AS is multifactorial in origin. The causes of bone loss in AS have long been speculated, if not always understood. The major pathogenetic factors of bone loss are systemic inflammation and decreased mobility of the spine.
Potential causes of osteoporosis in AS
1. Systemic inflammation
2. Poor spinal mobility
3. Low peak bone mass
4. Low physical activity
5. Life style factors such as smoking and alcoholism
6. Low BMI
7. Hypogonadism
8. Abnormal vitamin D metabolism
9. Malabsorption
10. Genetic factors
11. Abnormal bone turnover
12. Drugs: corticosteroids
13. Co morbid illnesses such as inflammatory bowel disease and psoriasis
14. Decreased skeletal loading
Systemic inflammation is likely the major contributing factor of bone loss in AS. This is because patients with AS have low BMD at both lumbar spine and the hip and spinal immobility cannot explain bone loss at the hip. Moreover, even patients with mild disease who are ambulatory and have normal spinal mobility develop low bone density (Bhalla 1992). The fact that even patients with early AS experience bone loss further suggests that systemic inflammation is the most likely explanation for bone loss in AS. Will and colleagues were the first to report that even patients with normal or mild involvement of the spine, normal mobility and early AS had low bone density (Will 1989). This notion has been confirmed by several recent studies (van der Weijden 2011, Maillefert 2001). A systematic review conducted by vander Weijden and colleagues reported that even patients within ten years of onset of AS developed bone loss (van der Weijden 2011). This review included seven studies with a total of 482 patients with AS.
The subjects were relatively young with a mean age of 35 years and had disease duration of 8 years. The prevalence of osteopenia vs. osteoporosis for lumbar spine was 39 vs. 16 % and for femoral neck, 38 vs. 13 % and this was similar to the prevalence rates in those with long-standing AS patients. Recently, a prospective study showed that bone loss was clearly due to inflammatory activity in early AS and not due to poor vertebral mobility and impaired daily physical activity (Gratacoal 1999). Further, the results of Mitra and colleagues suggest that bone loss is due to inflammation, because the cohort did not have advanced spinal disease, syndesmophytes limiting spinal mobility (Mitra 2000). Maillifert et al observed that patients with persistent systemic inflammation experienced significant bone loss over two years suggesting that inflammation is an important etiology of bone loss (Mallifert 2001). In this longitudinal study, the bone loss at femoral neck was related to persistent systemic inflammation, defined using ESR (mean percentage change –4.1% +5.7 and –1.2% + 3.9 in patients with and without persistent inflammation; respectively; p = 0.007). Data on the adverse effect of inflammation on BMD in patients with AS is fairly consistent (Grazio 2012, Arends 2011, Gratacoal 1999,Ghozlani 2009, Lange 2005, Toussirot 1999).
This argument is further supported by the various recent longitudinal studies that have noted that treatment with TNFi likely improve BMD at lumbar spine for up to six years by reducing inflammation (Durnez 2013,Kang 2011,Saad 2011, Kang 2013). TNFi inhibitors have also been shown to stabilize or improve BMD at the hip. The longitudinal gain in BMD noted at the lumbar spine was found to persist even in those without radiological changes in the spine proposing that the change in BMD reflected a true gain (Kang 2013). In a prospective longitudinal study conducted by Kang et al. in 26 patients, treatment with TNFi resulted in consistent gain in BMD at the lumbar spine and total proximal femur over two years compared to 37 patients not receiving TNFi (p < 0.01 and p = 0.02) (Kang 2013). Though mSASSS score, an indicator of radiological severity and progression had increased in those receiving TNFi, development of syndesmophytes was not different between the two groups. The authors also observed that treatment with TNFi inhibitors and the increase in SASSS were independently associated with increase in BMD at the lumbar spine BMD (p = 0.009 and p < 0.001). This further strengthens the observation that control of systemic inflammation likely improves BMD at lumbar spine and hip.
Studies have reported that bone loss in AS is linked partly to poor spinal mobility. Grazio and colleagues observed a significant negative correlation of BMD with serum markers of inflammation such as CRP (13+ 18 mg/L) and ESR (22+ 21 mm/h) (Grazio 2012). The negative association was more pronounced at hip than at the lumbar spine
Thus, it is evident that factors such as poor mobility as indicated by high BASMI contribute to bone loss in AS. Physical factors such as low body weight and lack of physical exercise may contribute to low BMD. El Maghraoui and Klingberg et al. have also observed the negative effect of low BMI on bone health in AS (El Maghraoui 1999, Klingberg 2012). Lack of exercise or regular physical activity is a frequent problem in patients with AS (Passalent 2010). A questionnaire-based study on 61 patients with AS, documented that walking and stretching were the most commonly followed types of exercise. However, these were reported only in 35.0% and 33% of patients respectively and most patients did not report doing exercise on a regular basis.
Presence of persistent pain, fatigue, stiffness of the back and spinal immobility limits physical activity. Progressive rigidity of the spine may cause changes in gait and stooped position and consequently impairing skeletal loading and BMD. Systemic bone loss is also mediated by abnormalities in endocrine pathways. Hypogonadism isan important risk factor for bone loss. Gonadal status may mediate bone loss in AS. Mitra et al. first studied the effect of hypogonadism on BMD in 1999 (Mitra 2000).
Similarly women with osteoporosis had significantly lower serum levels of estradiol (23.5 ± 34.4 vs. 80.9 ± 72.2 ng/l, p < 0.01) than those without osteoporosis. These results were replicated in a study done by Aydin and colleagues as well (Aydin 2005). In this cross sectional study of 58 young men (mean age: 38.2 years) with AS, serum DHEAS levels were low in 31% of patients with low BMD. The ratio of serum testosterone/DHEAS was higher in those with bone loss than those without (5·24 ± 3·70 vs. 3·58 ± 3·16, p = 0·026). But whether gonadal status is affected adversely in AS is still a matter of debate. Elevated DHEAS may be a response to systemic inflammation. In a small cross sectional study conducted in 2004, male patients (n=29) with AS were found to have higher levels of serum testosterone, LH, and prolactin than controls (El Maghraoui 2005). Thus, the potential role of androgens in initiating bone- loss in AS needs to be explored further.
Another crucial hormone that maintains skeletal health is vitamin D. Low vitamin D contributes to osteoporosis in AS in two different ways. First, vitamin D may have an aetio pathogenetic role in AS and low vitamin D may worsen inflammation. This is mainly because, 1, 25(OH)2D3, the active form of vitamin D, suppresses the proliferation of activated T cells and acts as an endogenous immune modulator (Amento 1987). Consequently, low levels of 1, 25(OH)2D3 may perpetuate the inflammation process in AS. Systemic inflammation affects bone turnover in a negative manner and contributes to bone loss. Second, low vitamin D levels cause bone loss by causing mineralization defects. Moreover, abnormalities of vitamin D can accelerate bone loss in AS (Arends 2011). There are several possible mechanisms by which patients with AS can have low vitamin D levels in serum. Firstly, patients with AS are known to have subclinical inflammation of the gut (Ebert 2004). Gut inflammation might result in poor absorption of nutrients including vitamin D. Secondly; serum levels of TNF-α remain high in AS patients that have active disease. TNF-α is known to down-regulate the activity of 1,25-hydroxylase enzyme in the kidney thus impairing the renal production of 1,25 vitamin D (Ebert 2004). It is also known that high disease activity in AS is associated with altered vitamin D metabolism and increased bone resorption (Lange 2001). Patients with active disease were found to have low vitamin D levels in a study done by Lange and colleagues in 2001 (78).
An inverse correlation was noted to exist between serum levels of 1,25(OH)2D3 (p<0.01) with disease activity. In addition, 1,25(OH)2D3 levels in serum correlated negatively with the excretion of urinary pyridinium crosslinks and positively with bone specific alkaline phosphatase (p<0.01). They also showed that AS patients with osteoporosis had significantly lower vitamin D levels compared to AS patients with normal BMD. In a previous study,patients with AS had lower mean 25-(OH) D3 levels (21.70 ± 12.17 vs. 32.70 ± 8.77 mmol/l) than healthy controls (Mermerci 2010). Though no difference was found between patients with or without osteoporosis groups in terms of serum 25-(OH)D3 levels, serum vitamin D levels were found to have an inverse association with serum CTX suggesting an influence on bone turnover (Mermerci 2010). Furthermore, data from a recent cross sectional study suggested that patients with active AS (n=100) had lower levels of 25-OH-vitamin D in serum (21.7 ± 12.2 mmol/l vs. 32.7 ± 8.8 mmol/l in controls, p= 0.0001) when compared to controls (Obermayer- Pietsch 2003). In a cross sectional study done in Dutch AS patients, median 25-OH-vitamin D level was 61.4 (13.8–186) nmol and serum 25OHvitD levels were independently related to sCTX suggesting a role in mediating bone loss (Arends 2011). Therefore it is clear that high disease activity in AS is associated with an alteration in vitamin D metabolism and increased bone turnover. Finally, some authors have also reported that AS is associated with a vitamin D receptor defect in the gut. In a study by Obermayer and colleagues in male AS patients, FokI genotypes of vitamin D receptor gene (VDR) was as independent predictor of low BMD at the spine (Obermayer-Pietsch 2003). Moreover CRP, ESR values were also significantly associated with FokI genotypes (total n=104, mean age 41+12 years). Thus, the VDR gene may be a risk factor for low BMD, abnormal bone metabolism and inflammation in AS.
It is also important to recognize that bone loss occurring from systemic inflammation is likely to be worsened by multiple confounding factors. These include poor peak bone mess, genetic factors, smoking, alcoholism, IBD and malabsorption occurring secondary to clinical or subclinical inflammation of gut.
Bone turnover in AS
Systemic inflammation exerts negative effects on bone turnover. Specifically, inflammation is associated with increased bone resorption and there are some reports suggesting that inflammation is also linked to reduced bone formation. Inflammation causes release of TNF alpha, IL-1 and IL -6, which then stimulate osteoclasts to cause bone resorption (Park 2008). Interestingly, AS is an inflammatory disease where both excess in bone resorption and new bone formation coexist (Arends 2011, Mitra 2000, Franck 2004, Lange 2001, Acebes 1999). A cross sectional study by Arends and colleagues found that serum P1NP, CTX and OC Z-scores were related to low BMD suggesting that AS is related to high bone turnover i.e. increased bone resorption and increased bone formation (Arends 2011).
Higher PINP, osteocalcin and sCTX Z-scores were significantly associated with low BMD at lumbar spine and hip in the regression analysis. In an earlier study done by Maghroui and colleagues, bone resorption markers such as urinary D-pyridinoline and C-telopeptide concentrations were increased in more than half of patients (53.9%). Also, the bone turnover markers positively correlated with CRP concentration suggesting that osteoporosis in AS occurred in parallel with inflammation (El Maghraoui 1999). Similar results were observed in another study in which serum ESR and NTX levels were significantly higher in patients with active AS (Borman 2008). Moreover, patients with active AS had significantly lower BMD and higher NTX (p < 0.05) levels (Borman 2008). In 2005, Lange and colleagues reported negative correlations between disease activity and serum levels of 1,25(OH)2D3 (p<0.01) and PTH (p<0.01) in a study conducted in 70 AS patients. Disease activity was expressed as high ESR,CRP and BASDAI (Lange 2001). In addition, they reported that the excretion of urinary pyridinium crosslinks showed a positive correlation with disease activity (p<0.01), and these results indicate that high disease activity in AS is associated with increased bone resorption (Lange 2001). Some studies have found a higher level of bone resorption markers in AS versus controls (83-86). For instance, Marhoffer et al. reported that osteocalcin concentrations were normal and similar to that of controls, but urinary pyridinium crosslinks were significantly increased in AS patients than with controls (51.2+ 25.2 vs. 33.9+12.4 in controls (p < 0.001)).
Serum ESR and CRP correlated only to bone resorption markers. These results were further confirmed by a study conducted by Briot and colleagues in 2005 (Briot 2005). Serum CTX levels increased and remained parallel to the increase in lumbar spine and hip BMD in patients receiving treatment with TNFi (Briot 2005). PINP levels in serum remained stable until 1 year despite an increasing until three months of treatment with TNFi (Briot 2005). BALP, OC and PINP are the main markers of bone formation. Most studies analyzing the serum levels of markers of bone formation in AS could not detect any differences in patients and controls (Park 2008, Acebes 1999, Marhoffer 1995, Yilmaz 2000, Vosse 2008, Toussirot 1999, Allali 2003, Woo 2007). However, in a study done by Mitra and colleagues, mean serum OC levels were lower in patients with AS than controls (9.0 vs. 11.0 ng/ml, p < 0.001) (Mitra 1999) Conversely, patients with AS had higher BALP levels in serum (mean: 38.5 vs. 30.3 U/l, p < 0.001) (Mitra 1999).
Furthermore, several studies have found a correlation between bone markers such as osteocalcin, uDPD and CTX with ESR and CRP suggesting a potential role for systemic inflammation as the cause for high bone turnover in AS (Toussirot 1999, Acebes 1999, Marhoffer 1995, Allali 2003, Mitra 1999).
Bone markers in relation to treatment with TNF alpha Inhibitors
Emerging data suggest that TNFi increase bone formation and decrease bone resorption in patients with AS. However, the data is inconsistent. Serum osteocalcin levels were found to increase at 6 weeks and then remain stable for up to 6 months after treatment with TNFi (Woo 2007). Likewise PINP levels in serum were shown to have an increasing trend at 3 months after infliximab treatment (Briot 2008). Similarly, Allali and colleagues studied 29 patients who were treated with infliximab and reported that there was an increase in serum osteocalcin levels (median: 23.8 mg/l) at 6 weeks (median change: 1.45 mg/l). However, the bone resorption marker urinary pyridinium did not change even after 6 months of treatment.
Recently, Arends et al. published a prospective cohort study of 111 AS patients who weretreated with TNFi for three years and their results suggested a rise in bone-specific alkaline phosphatase after 3 months (p<0.001) and the levels remained at a higher level up to three years. Similarly the serum levels of the bone formation marker PINP was found to be increased at three months and remained stable for up to 24 months (Arends 2012). Serum CTX levels decreased and remained decreased during three years of treatment with TNFi. In another study, 26 Korean patients receiving etanercept had a significant rise in serum levels of BALP and osteocalcin after 12 weeks of treatment (p < 0.05). But serum levels of CTX did not change after treatment with etanercept (Woo 2007). Although a difference was noted in the response of bone turnover markers between infliximab and etanercept, it is still unclear if the different types of TNFi affect bone metabolism differently.
RANKL and OPG system in AS
The role of the signaling pathway OPG/RANK/RANKL in mediating bone loss is now supported by many studies. Bone remodeling is modulated by two groups of cells namely osteoblasts and osteoclasts (Redlich 2012, Findlay 2011). The receptor activator of nuclear factor-κB (RANK) is a cell surface receptor present on the osteoclasts. The receptor activator of nuclear factor-κB ligand (RANKL) is a crucial regulator of the formation and function of osteoclasts and osteoprotegrin (OPG) is a negative inhibitor of osteoclastogenesis.
OPG is a decoy receptor that can inhibit the interaction between RANKL and its receptor RANK. The metabolic actions of RANKL and MMP are regulated by OPG a decoy receptor and TIMP respectively. There seems to be an important role for the RANKL-OPG system in AS. The tight balance between bone formation and bone resorption is impaired by inflammation. The inflammatory cytokines such as TNF-α and IL- 6 up regulate soluble RANKL, a mediator of osteoclastic bone resorption (Im 2009, Aydin 2005). In addition, inflammation leads to the release of bone and cartilage degrading enzymes such as cathepsin K and MMPs (Redlich 2012, Findlay 2011).
It is very important to better delineate how AS affects the RANK-L system. If the RANK-L system is proven to be more selectively affected in AS, targeted drugs such as denosumab can successfully be used to manage bone loss in patients with AS. Unfortunately, not much is known about how AS affects the OPG-the RANKL levels. Five studies have found that RANKL levels in serum are higher in patients with AS compared to controls (Kim 2006,Aydin 2005, Chen 2010, Taylan 2012, Rauner 2008). Existing data on this regard has shown heterogeneous results. In human studies, RANKL and RANKL/OPG ratio have been shown to be higher in AS patients with low BMD and this suggests a possible causative role of increased bone resorption in bone loss (Kim 2006, Taylan 2012). In a cross sectional study of 55 AS patients (38 had active AS) and 33 healthy controls, the RANKL/OPG ratio was higher in AS patients (.99 (0.14-22.6) vs. 0.65 (0.14-25.1), p=0.04, vs. controls). Another study found that HLA B27 transgenic rat models of SpA had higher mRNA levels of RANKL and RANKL/OPG ratio in bone tissue (Rauner 2009). However, other researchers have not replicated these results. A study conducted using surgical specimens of spinal tissue obtained from patients with AS failed to demonstrate increased sRANKL expression (Neidhart 2009).
Similarly, the results of a recent cross sectional study done by Dhir and colleagues from India in 85 patients (men (mean age: 33 ± 10 years and disease duration: 11 ± 7 years) showed that sRANKL (349.2 ± 872.0, 554.7 ± 1850.1, p = ns) did not differ between cases and controls and implied that sRANKL is not overexpressed in AS (Dhir 2013). Similarly, in a small study done by Woo J and colleagues on 26 AS patients receiving treatment with etanercept, sRANKL and OPG did not change after 12 weeks (Woo 2007).
Osteoprotegerin levels in AS
Researchers have reported contrasting results about how OPG is affected in AS. OPG is a soluble decoy receptor for RANKL and neutralizes the ability of RANKL to bind with RANK. OPG is also a key regulator of bone remodeling and the immune system. It can initiate osteoclastogenesis and also mediate communication between T cells with dendritic cells. Some studies have suggested that serum OPG levels are high in AS patients than in controls (Dhir 2013, Grisar 2002, Golmia 2002, Vandooren 2008). Also, a high OPG expression was demonstrated in synovial lining cells and endothelial cells in a study based on immunohistochemistry of patients with spondyloarthropathy (Haynes 2003). The increased production of OPG in AS could be a mechanism meant to compensate for bone loss and overproduction of RANKL. These results were not confirmed as some studies have found no difference or even lower levels of OPG in AS patients when compared to controls (Kim 2006, Taylan 2012). For instance, cross sectional data from a Korean study (n=60, 85% men and 15% premenopausal women) showed that serum levels of OPG in AS patients were not different (928 vs. 1158 pg/ml, p > 0.05) from controls (Kim 2006). Moreover, in 2004 Franck et al. observed that serum OPG levels were significantly lower in German patients with AS than controls.
WNT pathway in AS
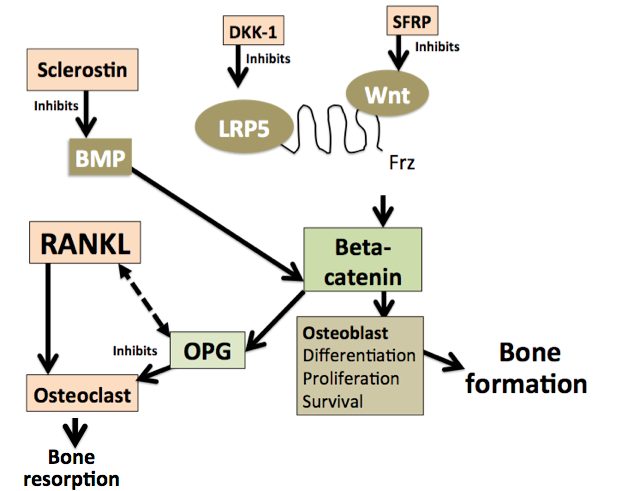
The Wnt-β-catenin pathway plays a crucial role in regulating bone turnover and metabolism. As shown in Figure 1, bone formation is regulated by BMPs, Wnt signaling and the Wntinhibitors such as DKK-1 and sclerostin. The BMP family modulates the initial part of the bone remodeling process whereas WNT proteins get involved only in the later stages. TNF alpha induced inflammation has the ability to modulate Wnt-β-catenin pathway.
Figure 1: Wnt signaling pathway
Data now suggests a possible role of the Wnt-β-catenin pathway in the pathogenesis and radiological progression of AS (Korkosz 2013). It can hence be argued that bone loss in AS is partly mediated by abnormalities of Wnt pathway. The Wnt -β-catenin pathway may be playing a dual role by mediating abnormal new bone formation and systemic osteoporosis in AS.Whether Wnt signaling is directly affected by AS or is it a consequence of systemic inflammation needs to be studied further.
Role of DKK-1 in AS
Sclerostin and functional Dkk1 are now being increasingly studied for their role in radiological progression in patients with AS. Serum DKK-1 levels have been shown to be high, low or unaffected in patients with AS (Taylan 2012,Korkosz 2013, Kwon 2012, Daoussis 2010). In a study done by Daoussis and colleagues, serum Dkk-1 levels were significantly increased in patients with AS (2,730 ± 135.1 pg/ml) as compared with normal controls (p = 0.040), patients with rheumatoid arthritis (p = 0.020), and patients with psoriatic arthritis (P = 0.049) (Daoussis 2010). Also, patients with AS receiving anti-TNF alpha treatment had significantly higher serum Dkk-1 levels than patients with AS who were not receiving such treatment (p = 0.007). Interestingly, Heiland and colleagues demonstrated that serum Dkk1 levels were significantly (p=0.025) higher in patients without syndesmophytes (6.78±5.48 pg/ml) compared with those with syndesmophytes (4.13±2.10 pg/ml) (Heiland 2012).
In the study by Taylan and colleagues, serum levels of various mediators of the Wnt pathway including DKK-1, sclerostin, and sFRP1, were not different in AS patients from the healthy controls (Taylan 2012). However, some patients studied (23/55) were already receiving treatment with TNFi and this might have influenced their results. Similar to the results of Daoussis and colleagues, in the study by Taylan and colleagues, patients receiving treatment with TNFi had higher DKK-1 levels compared to those taking conventional drugs (Daoussis 2010, Taylan 2012). Conversely, two later studies showed that serum DKK-1 levels either decreased or remained the same after using TNFi for 3 -6 months (Korkosz 2013, Kwon 2012). Two studies have also reported the presence of lower DKK-1 levels in AS patients than controls (Korkosz 2013, Kwon 2012). Serum DKK-1 level was lower in patients with AS (n=49) than in healthy controls (n=39) (12,321 + 6,136 vs. 20811 + 5,671 pg/ml, p<0.0001) (Kwon 2012). Furthermore, serum DKK-1 levels were increased in AS patients with low disease activity (Korkosz 2013). This may be the result of the compensatory response to the Wnt signaling being attenuated with low activity of inflammation in those with inactive disease (Korkosz 2013). Lower DKK-1 levels might promote bone formation and hence DKK-1 is currently being explored for its potential role in mediating new bone formation in AS. So far,studies have failed to demonstrate correlations between Dkk-1 and CRP (Taylan 2012, Korkosz 2013) in AS. Two studies reported no differences in serum DKK-1 levels in patients with active disease (BASDAI> 4) when compared to those with BASDAI <4 (Taylan 2012, Korkosz 2013).
Sclerostin abnormalities in AS
Osteocytes constitute the most abundant type of cells in bone. Sclerostin is an important product of osteocytes. It is a glycoprotein inhibitor of BMP and bone formation. n inflammatory arthritis, high TNF alpha has been found to induce skeletal expression of Dkk- 1 and sclerostin production, thereby inhibiting bone formation (Saad 2012, Daoussis 2010, Heiland 2012, Heiland 2010). Low sclerostin is also thought to be a marker of syndesmophyte formation in AS (Heiland 2012). In a recent study that was done to assess the sclerostin expression and osteocyte death in joints derived from patients with AS, the sclerostin expression was noted to be suppressed in patients with AS (14.7 ± 1.1%) when compared to those with rheumatoid arthritis, osteoarthritis and healthy controls (57.7 ± 3.2%, 42.2 ± 1.0% and 53.2 ± 3.1% respectively) (Heiland 2012). Also, serum levels of sclerostin were significantly lower in patients with AS than in healthy controls (326 ± 243 pg/ml in men; 81 ± 32 pg/ml in women) than in healthy control subjects (645 ± 221 pg/ml in men; 284 ± 152 pg/ml in women). Furthermore, the serum level of sclerostin over time was significantly higher in AS patients without syndesmophytes than in AS patients with syndesmophytes (p = 0.007).
However, not all studies have reported that sclerostin levels are low in AS. In 2013, cross sectional data presented by Korkosz and colleagues (n=50) suggested that the sclerostin levels in serum were significantly greater in patients with active disease (defined as BASDAI>4) than in controls (Korkosz 2013). In the study by Korkosz et al, there was no correlation between serum sclerostin or DKK-1 with CRP suggesting that molecular mechanisms other than inflammation might also be modulating Dkk-1 and sclerostin levels in AS. Conversely, in another study, the sclerostin levels were lower in patients receiving TNFi indicating that those with active and severe disease with high inflammation have higher sclerostin levels (Saad 2012). In this longitudinal study, at baseline, patients with AS had lower sclerostin levels (60.5 ± 32.7 vs. 96.7 ± 52.9 pmol/L, p = 0.002) compared to healthy controls. There were no differences in sclerostin levels between AS patients with or without peripheral joint involvement (69.9 ± 29.6 vs. 55.7 ± 33.9 pmol/L, p = 0.269) and between AS patients with a disease duration of > 5 years or < 5 years (48.13 ± 33.50 vs. 65.76 ± 31.71 pmol/L, p = 0.180).
Serum sclerostin levels showed inverse correlations with CRP (r = -0.57, p = 0.001) and ESR (r = -0.59, p < 0.001) suggesting that suppression of sclerostin occurs in the presence of inflammation. Though the serum levels of sclerostin increased from baseline (vs. 6 months vs. 12 months) after treatment with TNFi (60.5 ± 32.7 vs. 67.1 ± 31.9 vs. 72.7 ± 32.3 pmol/L, p <0.001) respectively), they remained significantly lower in patients compared to controls (72.7 ± 32.3 vs. 96.70 ± 52.9 pmol/L, P = 0.038) at 12 months. Furthermore, serum sclerostin levels at 12 months were lower in patients with high CRP (CRP ≥ 5 mg/L) compared to those with normal levels (50.8 ± 16.6 vs. 83.6 ± 33.0 pmol/L, p = 0.021). Similarly, in a recent report serum sclerostin levels did not change with TNF alpha inhibitor treatment (Korkosz 2014). The authors attributed this to the shorter disease course of patients as WNT proteins are activated only in later stages of bone remodeling. Conversely, Taylan et al observed that there were no differences in Dkk-1 and sclerostin levels between AS patients and controls and also between AS groups with high or low BASDAI (Taylan 2012). However, the results might have been influenced by the use of TNFi in some patients (23 / 55 patients studied). Sclerostin levels have also been studied in relation to BMD in AS. Not much data exists on the link between sclerostin and BMD in AS. In the study by Saad et al, BMD at the lumbar spine increased after treatment TNFi, but no significant change was observed for hip BMD and the change in spine BMD was attributed partly to sclerostin levels remaining lower after one year of treatment with TNFi (Saad 2008). Tsui and colleagues recently demonstrated higher than normal levels of noggin and sclerostin-IgG immune complexes in AS sera (p<0.001) (Winkler 2004). This study concluded that higher levels of NOG and/or SOST-IgG immune complexes probably contribute to new bone formation in AS patients (Winkler 2004,Tsui 2013). The role of noggin and sclerostin-IgG immune complexes in mediating bone loss needs to be studied.