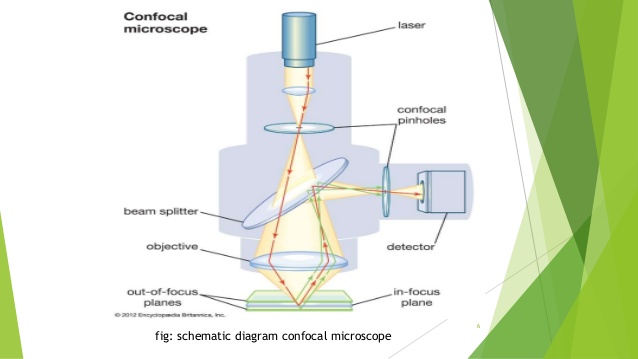
Confocal microscopy
Confocal microscopy is an optical imaging technique that enables 3D imaging of thick tissue. The schematic of a confocal microscope is shown in Figure 1. A point light source is used to illuminate the specimen; signal from the specimen is detected by a pinhole detector. The confocal schematic, i.e., the use of point source and point detector, enhances lateral resolution and more importantly, leads to depth discrimination capability. The point detector is in conjugation with the point source which is imaged to a slice of the specimen (i.e. point A and point P are in conjugation). The pinhole detector effectively places a small “aperture” extremely close to the object. Therefore, the lateral resolution is determined by the size of the virtue aperture instead of diffraction. This also decreases the field of view to infinitely small. However, the field of view can achieve a required size through mechanically scanning the focused light spot. Figure 1 also demonstrates the optical sectioning capability. If light gets reflected from the focal plane of the objective lens, the point A will be projected to the plane P as point B, where the point detector lies; therefore the reflected signal will be detected. However, when light is not reflected from the focal plane of the objective, point A will has a blurred image at plane P. Due to the pinhole configuration, only the central part of light will be detected; and as a result the out-of-focus light is rejected
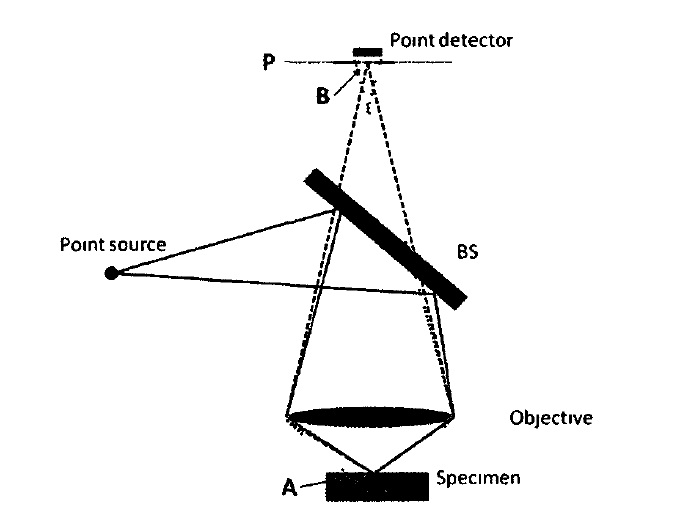
Figure 1 Schematic diagram of a confocal microscope
Two-photon excitation fluorescence microscopy
Similar to confocal microscopy, two-photon excitation fluorescence microscopy is also a widely used three-dimensional microscopic imaging technology Fluorescence excitation is the interaction between the fluorophore and an excitation electromagnetic field. Two-photon excitation involves the absorption of two photons whose combined energy is sufficient to induce a molecular transition to an excited state, as shown in Figure 2(a). As a comparison, Figure 2(b) shows the one-photon excitation. Since there are two photons involved, the excitation demonstrated in Figure 2 (a) is a nonlinear process and the excitation rate has a quadratic dependence on the excitation power. Through using a large NA microscope objective, the excitation will be confined to the focal plane of the objective. Besides the sectioning capability of two-photon excitation fluorescence microscopy, two-photon technique has improved contrast due to the large difference between excitation wavelength and emission wavelength For one-photon excitation, the excitation spectral band usually overlaps with the fluorescence emission band. A filter that rejects most excitation light may reduce background noise level; however, such a filter also rejects significant portion of fluorescence light to be detected A filter that allows most fluorescence light to be detected also allows a significant portion of excitation light to be detected. To the contrary, two-photon excitation has completely separated excitation spectrum and emission spectrum; therefore it is possible to use a filter to reject most of the excitation light and detect most of the fluorescence light. This can effectively improve the image contrast

Figure2 Jablonski diagrams for two-photon (a) and one-photon (b) excitation fluorescence